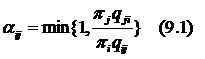1. Introduction
Inferential statistics give the basis to draw conclusions (or inferences) from the data and to evaluate the confidence in these conclusions being true. The need for inference is dictated by the fact that it is usually impossible to measure the parameter of interest in the whole population. The best alternative is to select a sample from the population and make the measurements. Then a conclusion may be derived about the population as a whole. However, due to a random chance, a sample may not represent the population well enough (i.e. most people in the sample may have higher than average resistance to the disease of interest). Therefore, the probability of such a coincidence has to be estimated as well.
The inferential statistics are generally considered a separate topic from descriptive statistics, which is mostly about the presentation of factual data.
2. Categories of Statistical Inference
2.1 Estimation
The population parameters can be estimated based on measuring the parameters of a sample and then calculating the mean and variance. These values can not tell what exactly the population parameters are. When a different sample is chosen, the values will differ slightly. Therefore, it is only possible to say that there is a certain confidence that the population parameters lie within some range. Generally, the accepted confidence for scientific studies is 95%.
2.2 Hypothesis testing
Hypothesis testing is used when there is a need to decide if some population values can be ruled out based on the result of the sampling. A hypothesis is a statement about the population. Hypothesis testing determines if there is enough evidence in the data to decide whether it is false. What is tested is called a null hypothesis, for example that the drug has no effect on the recovery from a disease. To achieve that, the probability is calculated that the data supports the null hypothesis, which is called p-value. The smaller the p-value, the stronger the evidence against a null hypothesis.
2.3 Decision theory
Decision theory attempts to combine the sample information with the other relevant aspects of the problem and make the best decision. Relevant information may include the consequences of a decision and a prior information that is available from sources other than the sample data. For example, a drug company may consider the decision about marketing a new painkiller. The ultimate decision will be whether to make a drug or not, including estimation of potential price, demand and profits.
3. Frequentist vs Bayesians
The basic definition of probability divides the statistics discipline into two distinct camps: frequentists and bayesians. A simple statement, such as "the probability that the coin lands heads is 1/2" can be viewed in very different ways. The frequentist will consider the following: if the experiment is repeated many times, the ratio of heads and tails will approach 1:1. So the probability is the long-run expected frequency. The bayesian will consider probability to be a measure of one's belief in the outcome.
In other words, frequentist does not know which way the world is. Also, since the data is finite, he can not tell exactly which way it is. So he will list the possibilities and see which ones are ruled out by the data. A bayesian also does not know which way the world is. He collects the finite data and compares how probable are different states of the world based on this data.
In some contexts frequentists and bayesians can coexist, but in some they are deeply in conflict. For example, to assign the numerical probability to the existence of life on Mars, we’ll have to drop the frequentist approach, because in frequentist point of view probability is something that’s determined in a lengthy series of trials.
This essay is written in terms of the frequentist approach.
4. Likelihood and Maximum Likelihood
Likelihood and probability are used as synonyms in everyday language, but from statistical point of view they are very different concepts. For example, the question about probability could be "what is the probability of getting two heads in a row". There is a parameter (P(heads)) and we estimate the probability of a sample given this parameter. The question about likelihood would be of a somewhat reversed direction. Is the coin fair? To what extent does the sample support the hypothesis that P(heads) = 0.5? We start with the observation and make inference about the parameter. In this example, the maximum likelihood of the coin being unfair is 1 as there is no data yet to support the "fair" hypothesis, while the probability of two head in a row is 0.5x0.5 = 0.25.
The likelihood function is defined as (1)
and is the joint probability of observing the data. The maximum likelihood is the estimate of a parameter that maximises the probability of observing the data given a specific model for the data.
Continuing the example with coin tossing, the model that describes the probability of observing head for a coin flip is called Bernoulli distribution. It has the form (2)
where p is the probability of heads and xi is the outcome of the ith coin flip.
If the likelihood function is applied to the Bernoulli distribution, the likelihood function becomes (3).
To simplify the equation, the logarithm of both sides can be taken, which does not change the value of the p for which the function is maximized (4).
Figure 1 shows the plots of likelihood as function of probability. As expected, the higher the proportion of one of the two outcomes, the more likely it is that the coin is not fair.
Figure 1 - Plots of likelihood as function of probability
The maximum likelihood as described above can also be determined analytically by taking the derivative of the likelihood function with respect to p and finding where the slope is zero. The maximum is found at (5).
which is just the proportion of heads observed in the experiment.
5. Regression
The simplest biological models are based on the concept of correlation. Correlation is observed when a change in one variable is observed together with the change in one or more other variables. For example, when the amount of calories consumed daily changes, other variables may change such as body weight or body fat percentage. Correlation is a measure of the relationship between these variables. It is important to keep in mind that a change in one of the variables does not necessarily cause the change in another. Both changes may, for example, be caused by some other, yet unknown, variable. For example, the length of the foot and the length of the palm are likely to correlate - people with large feet generally have large palms. But that does not mean that having large feet is caused by large palms, or the other way around. It is more likely that the overall body height is what causes the change in both variables.
Regression is a term that includes a number of different techniques that analyse the variables that are correlated and attempt to explore the relationship between those variables and the strength of correlation. The results may be used in developing models for prediction and forecasting.
For the regression analysis to be valid, some assumptions have to be satisfied:
- the sample is representative of the whole population
- the error is a random variable, so errors are independent of each other
- the errors are normally distributed
- the errors are not correlated
- the predictors, if there are more than one, are linearly independent
In the following discussion of linear and non-linear regression, the data gathered by Kuzmic et al (1996) is used. The data was gathered while studying asparatic proteinase catalysed hydrolysis of a fluorogenic peptide. The data is presented in Table 1.
| [S],μM | V0 |
|---|
| 0.66 | 0.0611 |
| 1 | 0.0870 |
| 1.5 | 0.1148 |
| 3 | 0.1407 |
| 4 | 0.1685 |
| 6 | 0.1796 |
Table 1. Experimental data
5.1 Linear Regression
The simplest form of regression is linear regression. It assumes that correlation between variables can be described by the equation y=ax+b, where x and y are the values of the variables. In this case, the relationship between variables can be plotted as a straight line. If the prediction about the linear nature of correlation is correct, and if the line is chosen well enough, the plotted values of variables will be located close to the "ideal" regression line. The problems here are first, to define the "closeness" of a variable to the predicted line and, second, to formulate a way to choose the line that will be the closest to all plotted values because there are an infinite number of possible lines and visual estimation is not at all precise enough.
The “closeness” is defined as the square of the vertical distance from the point to the regression line. The absolute value of the distance can not be used because it can be demonstrated that for any slope of the line it is possible to find such line that the absolute distances from points to the line, positive and negative, will cancel each other. Therefore, this does not solve the problem of finding a unique solution. If squared distance is used, however, the best line will go in the middle of two points, because the value of a2 + b2 is smallest when a = b.
Now that the value of the total squared error, which is the sum of squared distances from all points, is known, it is possible to choose which of the lines gives better fit to the data. The final task is to derive a method to predict which line will be the best fit.
The least squares estimates of slope and intercept are obtained as follows:
and
and
Or Sxy is the sum of x deviations times the sum of y deviations and Sxx is the sum of x deviations squared. Values of a and b are called the least squares estimates of slope and intercept.
As an example, it is possible to apply linear regression to the data from Table 1. That makes no practical sense since it is known beforehand that the correlation is not linear and is done only for illustration.
The data and the regression line can be plotted using the following R script
conc<-c(0.66,1,1.5,3,4,6)
rate1<-c(0.0611,0.0870,0.1148,0.1407,0.1685,0.1796)
plot(conc,rate1,lwd=2,col="red",xlab="[S], micromoles",ylab="V0")
par(new=TRUE)
abline(a=0.0683,b=0.0212,col="blue")
text(3, 0.12, "regression line", col="blue")
The results were presented in Figure 2.
Figure 2 - Linear regression against data from Table 1.
A null hypothesis can be examined, which states that V0 and S have no correlation. To achieve this, the Excel regression analysis was applied to determine the values of the standard error and the p-values. The result was presented in Figure 2.
Using the Excel function TINV(5%, 4) where 4 is the number of degrees of freedom and 5% means the 95% confidence, the value of the t statistic is 2.776. It is less that the critical value of 5.428, which allows to reject the null hypothesis and conclude that V0 correlates with S. This does not mean, of course, that the linear regression describes the correlation in the best way – there may exist a non-linear function that gives a better fit.
Another parameter describing the linear correlation is the correlation coefficient. It is a measure of the strength of the relationship of two variables and is defined as
Applying to the sample data,
and
The value of r is a number between -1 and 1, and the closer r is to 1, the stronger is the correlation, meaning that the values lie closer to the regression line. The value of r=0.9426 indicates that correlation is very strong and the linear regression produces a line that fits the data well. The lesson here is that it is easy to mistake a non-linear for a linear especially if there was no prior information about the nature of the relationship and the values are only measured in a limited interval.
5.2 Non-Linear Regression
Linear regression is a fairly simple technique and gives good results when the change in one variable has a constant influence on another. However, in biological and many other processes this is not always the case. As a simple example, as the length of a body increases, the mass increases too. But a specie which body length doubles will not double its mass - the increase in mass is likely to be greater than by the factor of two. To determine the nature of the relationship between variables when linear dependence does not apply, non-linear regression methods are used. Generally, there is no analytical solution for the non-linear least squares method.
In some cases, the non-linear functions can be transformed to the linear form. Consider the non-linear model of enzyme kinetics, the Michaelis-Menten equation (11)
The model can be transformed the following way:
Considering that the Km and Vmax are constants, the equation is now in the linear form of y=ax+b, where y=1/V, a=Km/Vmax and b=1/Vmax. The graphical representation of the function is known as Lineweaver-Burk plot and is used for the estimation of the Km and Vmax parameters. It is, however, very error prone because the y-axis takes a reciprocal of reaction rates and small measurement errors are increased.
When the analytical solution for the non-linear function does not exist, numerical methods are used. The algorithms behind these methods generally require an estimate for the parameter of interest, which should be reasonable to make sure the result is correct. In R language, nls function is used to determine nonlinear least-squares estimates of the parameters of a nonlinear model. To apply the function, it would be reasonable to first use the Lineweaver-Burk plot for an estimate of the parameters and then to use the results as an input for the nls function.
The reciprocal values were produced and the Lineweaver-Burk plot was constructed using the data from Table 1. Next, the least squares regression was applied to the plot of reciprocals to establish the values of Km and Vmax. This was achieved by the following R script
concLB<-1/conc
rate1LB<-1/rate1
fit<-lm(rate1LB~concLB)
fit
The script produced the following output.
Call:
lm(formula = rate1LB ~ concLB)
Coefficients:
(Intercept) concLB
4.051 7.853 The results were plotted by the following R script and are presented in Figure 3.
plot(concLB,rate1LB,ylim=range(c(rate1LB)),lwd=2,col="red",xlab="1/[S], 1/micromoles",ylab="1/V0")
par(new=TRUE)
abline(a=4.051,b=7.853,col="blue")
text(1, 10, "regression line", col="blue")
Figure 3-Using Lineweaver-Burk plot to estimate Michaelis-Menten parameters
The output was used to calculate the estimates for Km and Vmax.
1/Vmax = 4.051 + 7.853*(1/Km)Vmax = 1/4.051 = 0.2469-7.853*(1/Km) = 4.051-(1/Km)=0.5159Km = 1.9384Now that the reasonable estimates are calculated, it is possible to use the non-linear regression to fit Michaelis-Menten function. This is achieved by the following R script.
df<-data.frame(conc, rate1)
nlsfit <- nls(rate1~Vm*conc/(K+conc),data=df, start=list(K=1.9384, Vm=0.2469))
summary(nlsfit)
The script provides the following output
Formula: rate1 ~ Vm * conc/(K + conc)
Parameters:
Estimate Std. Error t value Pr(>|t|)
K 1.6842 0.2141 7.868 0.00141 **
Vm 0.2315 0.0112 20.664 3.24e-05 ***
---
Signif. codes: 0 *** 0.001 ** 0.01 * 0.05 . 0.1 1
Residual standard error: 0.005917 on 4 degrees of freedom
Number of iterations to convergence: 3
Achieved convergence tolerance: 2.895e-06
Alternatively, the R language contains a number of ‘self-starting’ models, which can be used without the initial estimates for the parameters. The Michaelis-Menten model is represented by SSmicmen function and can be used as in the following R script
model<-nls(rate1~SSmicmen(conc,a,b))
summary(model)
The script provides the following output
Formula: rate1 ~ SSmicmen(conc, a, b)
Parameters:
Estimate Std. Error t value Pr(>|t|)
a 0.2315 0.0112 20.664 3.24e-05 ***
b 1.6842 0.2141 7.868 0.00141 **
---
Signif. codes: 0 *** 0.001 ** 0.01 * 0.05 . 0.1 1
Residual standard error: 0.005917 on 4 degrees of freedom
Number of iterations to convergence: 0
Achieved convergence tolerance: 2.637e-06
In this case, the results were identical for the nls function and the SSmicmen function. This probably means that the initial estimates used in the nls function were reasonable. Lastly, the parameters identified by nonlinear regression can be used to plot the resulting function and compare it with experimental data.
The following R script will generate 100 points on the curve predicted by the values of Km and Vmax that were identified by the nonlinear least squares method and plot them against the experimental data.
plot(conc,rate1,lwd=2,col="red",xlab="[S], micromoles",ylab="V0")
par(new=TRUE)
Vm <- 0.2315
K <- 1.6842
x <- seq(0, 6, length=100)
y <- (Vm*x)/(K+x)
lines(x, y, lty="dotted", col="blue")
text(3, 0.13, "regression line", col="blue")
The output is presented in Figure 4.
Figure 4-Fitted curve plotted against experimental data.
To measure how well the fitted curve fits the data, a number of tests may be used. One of them is the F-test, which measures a null hypothesis that a predictor (in this case, [S], has no value in predicting V0. F is defined as (13)
where  is the ith predicted value,
is the ith predicted value,  is the ith observed value, and
is the ith observed value, and  is the mean. Specialized tables exist to obtain the critical values of F given the sample size and the required confidence percentage. When the F test is applied to two models, the test measures the probability that they have the same variance.
is the mean. Specialized tables exist to obtain the critical values of F given the sample size and the required confidence percentage. When the F test is applied to two models, the test measures the probability that they have the same variance.
Another test is the Akaike information criterion (AIC). It does not test the null hypothesis and, therefore, does not tell how well the model fits the data. It estimates how much information is lost when the attempt is made to fit the data to the model. In practice, it is used to select between several model the one that has the highest probability of being correct. If there are more than one model for which the AIC value is the same or very close, the best model may be a weighted sum of those models. AIC is defined as (14)
where k is the number of parameters in the model, and L is the maximised value of the likelihood function for the model.
References:
Aitken M, Broadhurst B, Hladky S,
Mathematics for Biological Scientists 2010, Garland Science, NY and London
Crawley, M,
The R Book, Wiley, 2007
Kuzmic P, Peranteau A, Garcia-Echeverria C, Rich D,
Mechanical Effects on the Kinetics of the HIV Proteinase Deactivation, Biochemical and Biophysical Research Communications 221, 313-317 (1996)
Ott R, Longnecker M,
An Introduction to Statistical Methods and Data Analysis, Brooks/Cole, 2010
Trosset M,
An Introduction to Statistical Inference and its Applications With R, Chapnam and Hall, 2009
Mathematics for Metabolic Modelling - Course Materials 2012, University of Manchester, UK
by Evgeny. Also posted on my website